Պարբերական հիվանդության և ամիլոիդոզի ախտածագումը, ձևաբանությունը, ախտորոշումը
Պարբերական հիվանդությունը (ՊՀ) կամ ընտանեկան միջերկրածովյան տենդը (ԸՄՏ), որը աուտոսոմ-ռեցեսիվ ժառանգվող հիվանդություն է, կլինիկորեն արտահայտվում է պարբերական տենդի նոպաներով, ուղեկցված շճաթաղանթների` որովայնամզի, թոքամզի, սինովիալ թաղանթների, ինչպես նաև սրտապարկի բորբոքումով և հաճախ` ամիլոիդոզի զարգացումով [1,3,5,19]: Այն էթնիկական նախատրամադրվածություն ունեցող հիվանդություն է, որով հիմնականում հիվանդանում են հայերը, սեֆարդ և աշքենազի հրեաները: Հիվանդությունը հանգեցնում է հաշմանդամության և աշխատունակության կորստի բավականին երիտասարդ տարիքում, հետևաբար, այն նաև կարևոր սոցիալական խնդիր է և շարունակում է մնալ որպես հայ բժշկագիտության արդիական և հրատապ խնդիրներից մեկը:
ՊՀ-ի զարգացումը պայմանավորող MEFV (Mediterranean fever) գենը տեղակայված է 16-րդ քրոմոսոմի կարճ թևում և կոդավորում է պիրին/մարենոստրին կոչվող սպիտակուցի սինթեզը, որը բաղկացած է 781 ամինաթթուներից [10,13]: Առաջարկված կարծիքների համաձայն, նրանով են հրահրվում հիվանդության նոպաները: Հայտնի է, որ պիրինը նորմայում, որպես միջնորդանյութ, վերահսկում է բորբոքային գործընթացը` կարգավորելով վերջինիս ադեկվատ արտահայտվածությունը: Այն արտադրվում է նեյտրոֆիլներում, էոզինոֆիլներում, մոնոցիտներում և դրսևորվում է որպես կասպազ-1-ի ակտիվացման կարգավորիչ: MEFV գենի մուտացիայի հետևանքով առաջացած փոփոխված պիրինն անընդունակ է դրսևորելու իր զսպիչ ազդեցությունը բորբոքային գործընթացում [7,14]:
ՊՀ-ի նոպաները բնորոշվում են տենդով, պոլիսերոզիտներով` պերիտոնիտով, պլևրիտով, և համեմատաբար հազվադեպ պերիկարդիտով` ուղեկցված որովայնային կամ կրծքավանդակի ցավերով, մոնոարտիկուլյար արթրիտով` ուղեկցված մասնավորապես խոշոր հոդերում հոդացավերով, մկանացավերով, ստորին վերջույթների` ոտնաթաթի և ներբանի մաշկի էրիթեմատոզ ցանով, ամորձու բորբոքումով: Որոշ դեպքերում նոպաներն ընթանում են անախտանիշ տենդի ձևով: ՊՀ-ով տառապող հիվանդների մեծամասնությունն իրենց առաջին նոպան ունեցել են մինչև 15-20 տարեկան հասակը: Միջնոպային շրջանում հիվանդները գործնականորեն առողջ են, թեև բորբոքային որոշ կենսաքիմիական և իմունաբանական ցուցանիշներ կարող են պահպանվել: Նոպաների հաճախականությունը խիստ տատանվում է հիվանդության ողջ ընթացքում: Դրանց մեծ հաճախականությունը` ամսվա ընթացքում երկու նոպայից ավելի, համարվում է պարբերական հիվանդության ընթացքի ծանրության չափանիշներից մեկը [3]: ՊՀ-ը հաճախ դրսևորվում է վաղ մանկական հասակում` սկսած կյանքի առաջին տարիներից: Հիվանդության վաղ դրսևորման, ինչպես նաև ծանր ընթացքի ռիսկի գործոններ են M694V, M680I և V726A մուտացիաները [6]: Ընդ որում, M694V մուտացիայի առկայության պայմաններում հիվանդությունը կարող է դրսևորվել կյանքի առաջին ամիսներից [2]:
ՊՀ սուր բորբոքային գրոհի դեպքում գերակշռում է նեյտրոֆիլների ներսփռումը դեպի շճաթաղանթներ, որը նրանց ուժեղացած արտագաղթի հետևանք է [14,15]: Շրջանառող նեյտրոֆիլներում էքսպրեսվող պիրինը հավելում է նշված բջիջների ակտիվացումը բորբոքային գործընթացի ժամանակ [7]:
Որպես ՊՀ հիմնական բարդություն, ամիլոիդոզի ձևավորման գործընթացում առանցքային նշանակություն ունի շիճուկային ամիլոիդային A-սպիտակուցի (SAA) մակարդակի բարձրացումն արյան մեջ (ավելի քան 1000մգ/լ): SAA սպիտակուցը սինթեզվում է հեպատոցիտներում, որը խթանվում է մակրոֆագային IL-1-ի ազդեցությամբ: SAA սպիտակուցի ինտենսիվ սինթեզի պայմաններում վերջինիս լրիվ դեգրադացիա մակրոֆագային համակարգի կողմից չի ապահովվում, և նրա ֆրագմենտներից ամիլոիդոբլաստներում տեղի է ունենում ամիլոիդային թելիկների կառուցում: Այս գործընթացը խթանվում է ամիլոիդ-խթանող գործոնի (AEF) միջնորդությամբ: Վերջինս ամիլոիդոգենեզի ընթացքում առաջացող նյութ է, որն արագացնում է ամիլոիդային կուտակումների ձևավորումը հյուսվածքներում [20]:
Փաստորեն, AA-ամիլոիդոգենեզում մակրոֆագային համակարգը կենտրոնական դեր ունի, քանզի այն խթանում է SAA սպիտակուցի սինթեզը լյարդում, և ինքն էլ մասնակցում է ամիլոիդային թելիկների կառուցմանը նրա ոչ լրիվ դեգրադացված ֆրագմենտներից [14]: SAA սպիտակուցի մոլեկուլի գենետիկորեն պայմանավորված կառուցվածքային շեղումները դիմադրողունակ են դարձնում վերջինիս մոնոցիտ/մակրոֆագերի տարրալուծիչ հատկության հանդեպ:
Ամիլոիդոբլաստների աղբյուր կարող են հանդիսանալ տարբեր օրգանների և հյուսվածքների մասնագիտացված մեզենքիմային բջիջների նախորդները` կախված օրգանի կամ հյուսվածքի կառուցվածքագործառական առանձնահատկություններից (երիկամում` մեզանգիոցիտները կամ էնդոթելային բջիջները, լյարդում` կուպֆերյան բջիջները, փայծաղում` ռետիկուլային բջիջները, սրտում` կարդիոմիոցիտները, անոթների պատում` հարթ մկանաբջիջները և այլն): Ամիլոիդի թելիկային բաղադրամասի ձևածագման գործընթացն իրականանում է 3 հաջորդական փուլերով` լյարդում SAA սպիտակուցի սինթեզի խթանում IL-1-ով, որը հանգեցնում է նախաամիլոիդային SAA սպիտակուցի մակարդակի բարձրացմանն արյան պլազմայում, նրա ոչ լրիվ ֆերմենտատիվ դեգրադացիա մակրոֆագերով ու լուծելի AA-սպիտակուցի գոյացում, և վերջինից ամիլոիդային թելիկների կառուցում ամիլոիդոբլաստներում` ամիլոիդ-խթանող գործոնի ազդեցությամբ [23]: Ամիլոիդային թելիկների ձևավորմանը զուգահեռ ինչ-որ չափով ընթանում է նաև նրանց տարրալուծում ամիլոիդոկլաստների միջոցով (ամիլոիդոկլազիա), որը սակայն թույլ է արտահայտված: Այն բացատրվում է ամիլոիդային սպիտակուցի հանդեպ հանդուրժողականության (տոլերանտության) ձևավորումով: Ամիլոիդային թելիկների հետ պլազմայի գլիկոպրոտեիդների, հյուսվածքային քոնդրոիտին սուլֆատի միացումն ավարտվում է բարդ գլիկոպրոտեիդ ամիլոիդի առաջացմամբ:
Ամիլոիդի ձևավորման մեխանիզմները դեռևս լիովին լուսաբանված չեն: Կան կարծիքներ, որ ամիլոիդի առաջացումն առանձնահատուկ է յուրաքանչյուր անհատի համար, կամ պայմանավորված է SAA-ի իզոտիպով: SAA սպիտակուցի հայտնի երեք իզոտիպերից SAA 1.5-ը պայմանավորում է ամիլոիդոզի առաջացման առավել նվազ հավանականություն` կայունություն ցուցաբերելով մատրիքսային մետալոպրոտեինազներով (հատկապես MMP-1-ով) դեգրադացիայի հանդեպ, ի տարբերություն SAA 1.1 և 1.3 իզոտիպերի, որոնք առավել անկայուն են դեգրադացիայի հանդեպ, դրանով իսկ նպաստելով ամիլոիդոզի առաջացմանը` հատկապես թելիկագոյացման գործընթացին, կամ դրսևորելով ամիլոիդ-խթանող գործոնին համանման ազդեցություն [23]:
SAA սպիտակուցի փոխակերպումը ամիլոիդի ուսումնասիրվել է մարդկային պերիտոնեալ բջիջների և արյան մոնոցիտների մոդելների վրա: Պարզվել է, որ մարդու մոնոցիտային կուլտուրան, ի տարբերություն պերիտոնեալ բջիջների, ավելի ակտիվ է ամիլոիդի ձևավորման գործում ամիլոիդ-խթանող գործոնի բացակայության պայմաններում: Ըստ հետազոտության արդյունքների, AA ամիլոիդային թելիկներն առաջանում են բջջի ներսում, թելիկների էկզոցիտոզն իրականանում է լիզոսոմներից ծագած վեզիկուլներով: Մարդկային մոնոցիտները ցուցաբերել են ամիլոիդոգենեզի նախնական փուլն իրագործելու ունակություն` SAA սպիտակուցի ներբջջային կուտակումով, թելիկների միակցումով, սկզբնավորվող ամիլոիդի տեղաբաշխումով դեպի արտաբջջային միջավայր, գլիկոզամինոլիկանների միավորումով ամիլոիդին [17]:
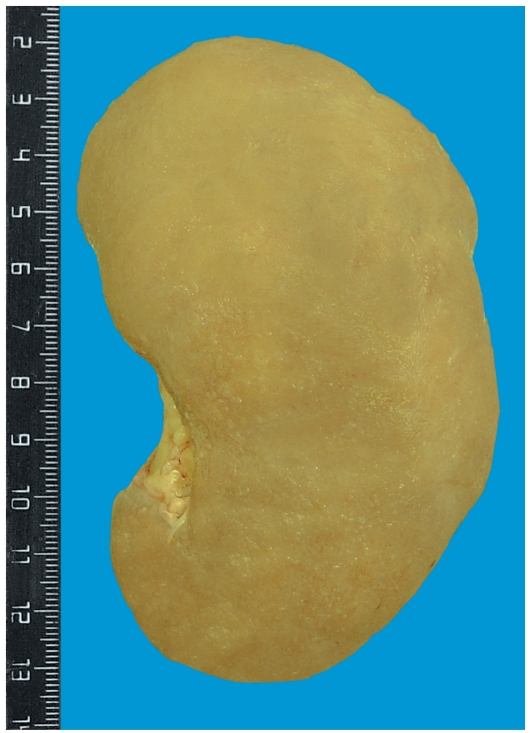
Համակարգային ամիլոիդոզը ներքին օրգաններում, որպես ՊՀ հիմնական բարդություն, բնորոշվում է ամիլոիդի առավել արտահայտված կուտակումներով երիկամներում, որն արտահայտվում է սպիտամիզությամբ և նեֆրոտիկ համախտանիշով, փայծաղում, որը սովորաբար անախտանիշ է ընթանում: Ավելի ուշ ամիլոիդային կուտակումներ հայտնաբերվում են լյարդում, աղիներում, սրտում և այլ օրգաններում: Երիկամային ամիլոիդոզը ՊՀ ամենահաճախ հանդիպող բարդությունն է, որը հանգեցնում է երիկամների ամիլոիդային կնճռոտման և երիկամային անբավարարության, որն էլ, ըստ գրականության և մեր կողմից կատարված դիահերձման տվյալների, ՊՀ հիվանդների մեծամասնության մահվան պատճառն է:
Երիկամների ամիլոիդային ախտահարումն արտահայտվում է երիկամների բրգերում` ուղիղ անոթների և հավաքող խողովակների ուղղությամբ, որ հայտնաբերվում է միկրոսկոպիորեն, այնուհետև կծիկներում` մեզանգիումում, էնդոթելի տակ, անոթների պատերում, խողովակների սեփական թիթեղի ուղղությամբ ամիլոիդ նյութի կուտակումներով: Տրանսֆորմացնող աճի գործոն-β-1-ը նշանակալիորեն նպաստում է AA երիկամային ամիլոիդոզի և երիկամների կեղևային շերտի միջանկյալ հյուսվածքի ֆիբրոզի զարգացմանը [8]: Կան տվյալներ, որ հղիությունը կարող է խթանել ՊՀ-ի հետ կապված ամիլոիդոգենեզը երիկամներում: Երիկամի ամիլոիդոզի հարաճուն զարգացման գործընթացում մեծ հակվածություն է ցուցաբերում M694V-ով հոմոզիգոտ գենոտիպը [6,21]:
ՊՀ-ով պայմանավորված ներզատիչ գեղձերի ամիլոիդային ախտահարումը կարող է արտահայտվել բազմագեղձային ներզատիչ անբավարարությամբ: Այն ընդգրկում է տարբեր ներզատիչ գեղձեր` մակերիկամներ, վահանագեղձ, հարվահանագեղձեր, ամորձիներ, սակայն նրանցում կլինիկորեն դրսևորվող գործառական բնույթի նշանակալի խանգարումներն ակնհայտ չեն: Այսպիսով` ներզատիչ անբավարարությունն ունի գաղտնի կամ նվազ ախտանշային բնույթ և հաճախ դրսևորվում է ամիլոիդային ախտահարումների խիստ արտահայտվածության դեպքում [12,18]:
ՊՀ-ով պայմանավորված սրտի ամիլոիդոզը, որպես խրոնիկական հարաճուն ախտահարում արտահայտվում է կարդիոմեգալիայով և ուղեկցվում սրտի հաղորդականության խանգարումներով: Այն հաճախ կլինիկորեն դրսևորվում է ՊՀ հիվանդի երիկամի հաջող փոխպատվաստումից տարիներ անց միայն` հանգեցնելով մահվան սրտային անբավարարությունից: Սակայն որոշ դեպքերում սրտային անբավարարությունը, որպես սրտի ամիլոիդային ախտահարման ելք, մահվան պատճառ կարող է դառնալ մինչև երիկամի ամիլոիդոզի հետևանքով առաջացած ուրեմիայի զարգացումը [24]: Հոգեհուզական և ֆիզիկական սթրեսը, ինչպես նաև M694V մուտացիայի հոմոզիգոտ վիճակը ՊՀ-ով պայմանավորված սրտի ամիլոիդային ախտահարման վճռորոշ ռիսկի գործոններ են [5]:
ՊՀ-ով պայմանավորված թոքային սփռուն ամիլոիդոզը հաճախ կլինիկորեն դրսևորվում է թոքաբորբի ձևով: Այն ուղեկցվում է թոքամզի թերթիկների միջև կպումների առաջացումով, թոքամզի սկլերոզով կամ ամիլոիդոզով: Թոքային ամիլոիդոզը, ըստ արտահայտված կարծիքների, սովորաբար կլինիկորեն արտահայտվում է թոքային հիպերտենզիայի ձևով, արդեն իսկ առկա երիկամային անբավարարության շրջանում [9,19]:
ՊՀ-ով հիվանդների արյունաբանական հետազոտությունը բացահայտել է ԴՆԹ-ի ռելիեֆության, տեքստուրայի և մասնիկների փոխդասավորվածության, կորիզակի տրամագծի փոփոխություններ` բնորոշ հատկապես ամիլոիդոզի զարգացմանը [4]: Կան կարծիքներ, որ ՊՀ-ի ախտորոշման կարևոր ցուցանիշ է լեյկոտրիեն B-4-ի (LTB4) բարձր մակարդակը մեզում [16]:
ՊՀ-ով պայմանավորված համակարգային ամիլոիդոզի ախտորոշման հիմնական եղանակը բիոպսիան է: Բիոպսիոն նյութի ուսումնասիրությունը հատուկ ներկման եղանակներով` կոնգո կարմիրով (լուսային և պոլյարիզացիոն մանրադիտակային հետազոտությամբ), զուգակցված իմունահիստոքիմիական մեթոդներով, կարևոր են ՊՀ-ով պայմանավորված ամիլոիդոզի հայտնաբերման համար:
Երիկամի բիոպսիան արդյունավետ է, քանի որ ՊՀ-ի ժամանակ այն առավել հաճախ է ախտահարվում ամիլոիդոզով: Երիկամային ամիլոիդոզի ախտորոշումը թիոֆլավին-T –ով հաջողված մեթոդ է համարվում: Նվազ տրավմատիկ և միևնույն ժամանակ ինֆորմատիվ է ուղիղ աղու բիոպսիան: ՊՀ պայմանավորված ամիլոիդոզը 12-մատնյա աղիում արտահայտվում է առավել, քան ստամոքսում, որտեղ էլ անտրալ բաժնի ամիլոիդոզն ավելի հաճախ է հանդիպում, քան ստամոքսի մարմնում [3]: Վահանագեղձի նույնիսկ լավ արտահայտված ամիլոիդային կուտակումների առկայության պայմաններում օրգանի ֆունկցիան կարող է էապես չտուժել: Վերջինիս ախտորոշման համար, որպես համեմատաբար անվնաս և հեշտ իրագործելի միջամտություն, կիրառվում է ասեղնային ասպիրացիոն բիոպսիոն եղանակը [18]:
Լյարդի ամիլոիդային ախտահարման դեպքում, որը բնորոշվում է հեպատոմեգալիայով, պունկցիոն ասպիրացիոն բիոպսիոն եղանակով հայտնաբերվում են ամիլոիդ նյութի կուտակումներ ճնշված, ապաճած հեպատոցիտների միջև, խրոնիկական բորբոքմանը բնորոշ բջջային ռեակցիա և չախտահարված լյարդային հյուսվածքի տեղամասեր [11]: ՊՀ-ով հիվանդի ամորձու բիոպսիոն հետազոտության միջոցով հայտնաբերվում են ամիլոիդ նյութի առատ կուտակումներ և սպերմատոգենեզից զուրկ, հիալինոզի ենթարկված խողովակներ [12]: Ենթամաշկային ճարպաբջջանքի ասպիրացիոն բիոպսիոն ախտորոշիչ մեթոդը ևս բավականին ինֆորմատիվ է և կիրառվում է ամիլոիդոզի հայտնաբերման համար [22]:
Ներկայումս խիստ արդիական է ՊՀ-ով պայմանավորված ամիլոիդոզի հնարավորինս վաղ ախտորոշման խնդիրը: Մեր կողմից մշակվում են ժամանակակից և նվազ տրավմատիկ մեթոդներ ամիլոիդոզի վաղ` գաղտնի շրջանում ախտորոշման համար:
Գրականություն
- 1. Айвазян А.А. Периодическая болезнь. Ереван, 1982, 215 с.
- 2. Амарян Г.Г. Факторы риска ранней манифестации и повышенной экспрессивности периодической болезни у детей. Медицина, наука и образование. Научно-информационный журнал, 2009, 3, с. 15-22.
- 3. Еганян Г.А. Клиническое значение и патогенез поражений желудочно-кишечного тракта при ПБ. Автореф. докт. дис. М., 1991, 37с.
- 4. Захарян А.Г. и соавт. Оценка морфофункционального состояния лимфоцитов при периодической болезни. Материалы международной научно-практической конференции, посв. 75-летию основания Гематологического центра им. проф. Р.О. Еоляна Ж. Кровь, 2008, 2 (8), с.68.
- 5. Назаретян Э.Е., Гаспарян А.Ю. Поражение сердца при ПБ. Мед. наука Армении, НАН РА, 2000, 1, с.53-56.
- 6. Саркисян Т.Ф., Айрапетян А.С., Шахсуварян Г.Р., Егиазарян А.Р. Молекулярная диагностика семейной средиземноморской лихорадки (ПБ) среди армян. Новый армянский медицинский журнал, 2007, т.1, 1, с. 1-8.
- 7. Chae J.J., Wood G., Masters S.L., Richard K., Park G., Smith B.J. et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc. Natl. Acad. Sci. U S A, 2006;103:9982-9987.
- 8. Danilewicz M., Wagrowska-Danilewier M. The role of transforming growth factor in development process of AA and AL kidney amyloidosis: according to results of immunohystochemical analises. Pol. J. Pathol., 2006; 57(4); p.193-8.
- 9. Erdem H., Simsek I., Pay S., Dinc A., Deniz O., Ozcan A. Diffuse pulmonary amyloidosis that mimics interstitial lung disease in a patient with familial Mediterranean fever. J. Clin. Rheumatol., 2006;12(1):34-36.
- The French FMF Consortium. A candidate gene for FMF. Nat. Genet., 1997, 17:25-31.
- 10. Gangane N., Anhu, Shivkumar V.B., Sharta S. Aspiration biopsy as a diagnostic method for hepatic amyloidosis. Acta Cytol., 2006; 50(5), p. 574-576.
- 11. Haimov-Kochman R., Prus D., Ben-Chetrit E. Azoospermia due to testicular amyloidosis in a patient with familial Mediterranean fever. Hum. Reprod., 2001, 16(6), p.1218-1220.
- 12. The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause FMF. Cell, 1997, 90:787-807.
- 13. Korkmaz C., Ozdogan H., Kasapcopur O., Yazici H. Acute phase response in Familial Mediterranian Fever. Ann. Rheum. Dis., 2002, 61, p.79-81.
- 14. Lechmann H.J., Sengul B., Yavuzsen T.U., Booth D.R., Booth S.E., Bybee A. Clinical and subclinical inflammation in patients with Familial Mediterranian Fever and in heterozygous carriers of MEFV mutations. Rheumatol., 2006, 45, p.746-750.
- 15. Livneh A., Langevitz P. Diagnostic and treatment concerns in familial Mediterranean fever. Baillieres Best Pract. Res. Clin. Rheumatol., 2000, 14 (3): 477–498.
- 16. Magy N., Kluve-Beckerman B., Benson M. Human monocytes have complete capacity to initiate pathogenesis of reactive amyloidosis. Meeting reports of the fourth international congress on systemic autoinflammatory diseases. National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), 2005.
- 17. Ozdemir B.H., Uyar P., Ozdemir F.N. Aspiration biopsy as a diagnostic method for thyroid amyloidosis. Cytopathology, 2006, 17(5), p.262-266.
- 18. Sahan C., Cengiz K. Pulmonary amyloidosis in familial Mediterranean fever. Acta Clin. Belg., 2006, 61(3):147-51.
- 19. Strasburg S., Pras M., Gal R., Salai M., Livneh A. Inhibition of the second phase of amyloidogenesis in a mouse model by a single-dose colchicines regimen. J. Lab. Clin. Med., 2001, 138, p.107-111.
- 20. Touitou I., Sarkisian T., Medlej-Hasim M., Tunca M., Livneh A. et al. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis & Rheum., 2007; 56 (11): 3879-3880.
- 21. Van Gameren I., Hazenberg B., Bijzet J., Van Rijswijk M. Diagnostic accuracy of subcutaneous abdominal fat aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum., 2006, 54: p.724-729.
- 22. Yakar S., Livneh A., Kaplan B., Pras M. The molecular basis of reactive amyloidosis. Seminars in Arthritis and Rheumatism, 2003, vol.24, I.4, p. 255-261.
- 23. Yildiz A., Akkaya V., Kilicaslan I., Turkmen A., Gorcin B., Atilgan D., Sever MS. Cardiac and intestinal amyloidosis in a renal transplant recipient with Familial Mediterranian fever. J. Nephrol., 2001, 14(2):125-127.
| Հեղինակ : | Ս.Վ. Համբարձումյան Երևանի Մ. Հերացու անվան պետական բժշկական համալսարան, ախտաբանական անատոմիայի ամբիոն |
| Աղբյուր : | Գիտա - գործնական Բժշկական Հանդես <<Մեդիցինսկիյ Վեստնիկ Էրեբունի>> - 1. 2010 (41) 17-22 |
| Ներկայացնող` | Doctors.am |