






 Բնորոշումը
Բնորոշումը
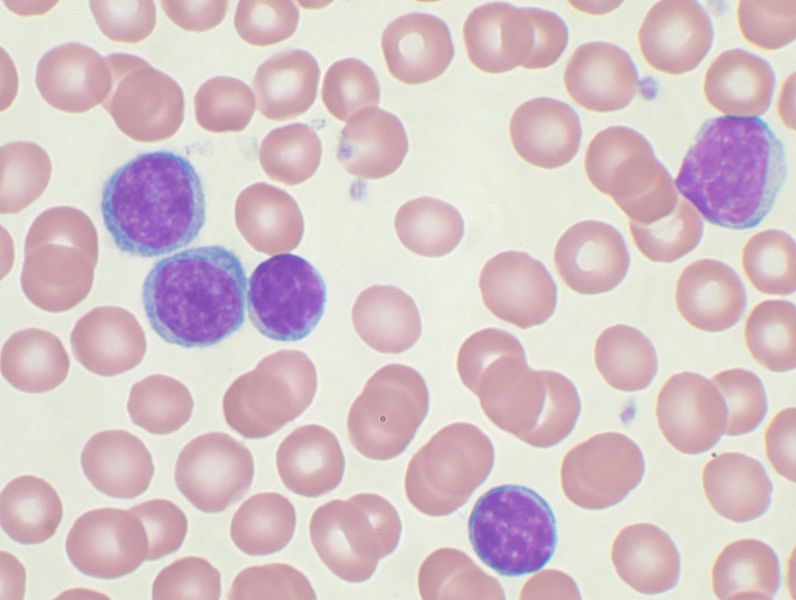
Քրոնիկական լիմֆոլեյկոզը (ՔԼԼ) կլոնալ նեոպլաստիկ լիմֆոպրոլիֆերատիվ հիվանդություն է, բնորոշվում է ոսկրածուծի լիմֆոիդ ներսփռումով, ծայրամասային արյան մեջ արտահայտված լիմֆոցիտոզով, լիմֆադենոպաթիայով, հեպատոսպլենոմեգալիայով:
Տարածվածությունը
Հիվանդությանը բնորոշ է ազգային և ռասայական տարբերությունը: Ասիական երկրներում հանդիպում է հազվադեպ: Բուրիաթների մոտ ոչ մի դեպք չի նկարագրված, հաճախ է հանդիպում հրեաների մոտ: Հիվանդությունը ԱՊՀ տարածքում կազմում է 0.4-4.0 £ 100000 բնակչությանը, Եվրոպայում և Ամերիկայում՝ 3.5 £ 100000: Տղամարդիկ երկու անգամ ավելի հաճախ են հիվանդանում, քան կանայք: Հիվանդությունը հիմնականում հանդիպում է 60 տարեկանից բարձր անձանց մոտ: Նկարագրված են ընտանեկան դեպքեր (հորիզոնական և ուղղահայաց):
Ծագումնաբանություն և ախտաբանություն
Կլինիկա
ՔԼԼ-ն հածախակի ունենում է անախտանշային ընթացք և հայտնաբերվում է պատահական՝ ծայրամասային արյան քննության արդյունքների հիման վրա: Անախտանշային շրջանը կարող է տևել տարիներ: Առհասարակ, հիվանդությունն իր բնույթով, զարգացման արագությունով շատ բազմազան է: Մի շարք հիվանդների մոտ հիվանդությունն ընթանում է ագրեսիվ և չնայած տարվող բուժմանը՝ տևում է 2-3 տարի: Այլ դեպքերում հիվանդությունն ընթանում է համեմատաբար հանգիստ, շնորհիվ տարվող բուժման ընդունում է դանդաղ, հարաճուն ընթացք:
Ըստ ընթացքի բաժանում են հետևյալ կլինիկական շրջանների
| 0 շրջան | ոսկրածուծում լիմֆոցիտոզ 40% | ծայրամասային արյան մեջ > 15,0ճ103/լ |
| I շրջան | լիմֆոցիտոզ | լիմֆադենոպաթիա |
| II շրջան | լիմֆոցիտոզ | սպլենոմեգալիա կամ/և հեպատոմեգալիա |
| III շրջան | լիմֆոցիտոզ | անեմիա (ոչ աուտոիմուն), լիմֆադենոպաթիա, օրգանոմեգալիա կարող է չլինել |
| IV շրջան | լիմֆոցիտոզ | թրոմբոցիտոպենիա (ոչ աուտոիմուն), անեմիա և օրգանոմեգալիա կարող են չլինել |
Հիվանդության ընթացքը 70-80% դեպքերում բարդանում է կրկնվող, ձգձգվող ինտերկուրենտ ինֆեկցիաներով, որոնք պայմանավորված են հիպոգամմագլոբուլինեմիայով, գրանուլոցիտոպենիայով և հաճախ հանդիսանում են հիվանդների մահվան հինական պատճառ: հիվանդների 10%-ի մոտ զարգանում են աուտոիմուն հեմոլիտիկ անեմիաներ և թրոմբոցիտոպենիաներ: ՔԼԼ-ը բավականին հաճախ զուգորդվում է երկրորային ուռուցքային հիվանդություններով (մելանոմա, սարկոմա, միելոմային հիվանդություն, թոքի և հաստ աղու քաղծկեղ), ամենից հաճախ՝ մաշկի ուռուցքով:
Բուժումը
ՔԼԼ բուժման ծրագրի ընտրությունը կախված է հիվանդության կլինիկական շրջանից՝ կամ մոնոթերապիա, կամ կոմբինացված քիմիաթերապիայի ծրագրեր: Օգտագործվող հիմնական դեղաձևերն են՝ լեյկերան (քլորբութին, քլորամբուցիլ), ցիկլոֆոսֆան (էնդոքսան), ֆլյուդարաբին, կորտիկոստերոիդներ, վերջին տարիներին նաև մոնոկլոնալ հակամարմիններ (անտի-CD 20՝ մաբտերա), ինտերֆերոն, ինտերլեյկին-2:
Էլեկտրոնային նյութի սկզբնաղբյուրը ՝
Երևանի Մ. Հերացու անվան պետական բժշկական համալսարան



